
Our children were handed a death sentence when a simple blood test at birth could have saved their lives – by the time doctors took us seriously, it was too late for treatment


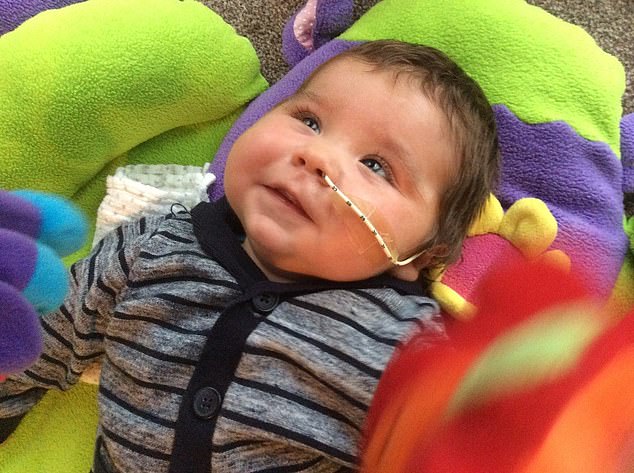




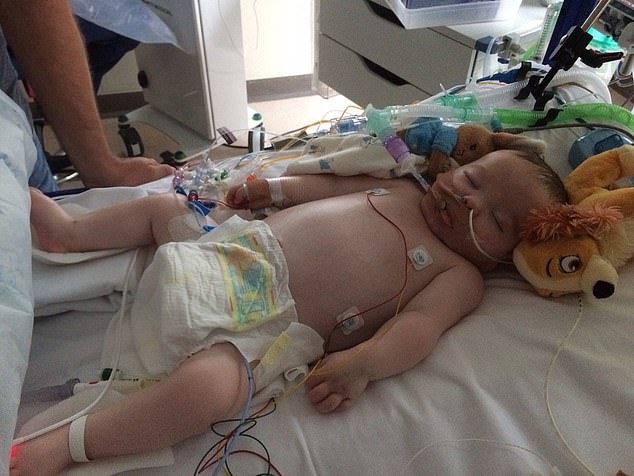




As their baby son James lay seriously ill on his hospital bed, his desperate parents could only look on helplessly as doctors battled to work out what was wrong.
‘At one point they thought it might be meningitis, then pneumonia, or maybe epilepsy,’ his mum, Susie Thorndyke, recalls.
‘Each of these things seemed so devastating – but little did we know.’
It would take another agonising few weeks before Susie, 41, and her husband Justin, 47, learned that three-month-old James — now intubated and in intensive care — had severe combined immunodeficiency (SCID), a genetic disorder that causes major abnormalities of the immune system.
Without extremely early intervention, children usually die within the first two years of their life.
‘We couldn’t quite believe what was happening,’ Susie says now. ‘It was surreal, devastating. Suddenly we were effectively watching our baby die.’
For while James did receive treatment in the form of a bone marrow transplant, it came too late.
His condition continued to deteriorate, and he passed away in February 2017, five days before what would have been his first birthday.

In 2016, Susie and Justin Thorndyke learned that three-month-old James had severe combined immunodeficiency (SCID), a genetic disorder that causes major abnormalities of the immune system

His condition continued to deteriorate, and he passed away in February 2017, five days before what would have been his first birthday
Desolating enough for Susie and Justin, from Norfolk, but there was also a final ‘twist of the knife’: had James received a simple blood test at birth — a specialised version of the routine heel prick test offered to all newborns.
The condition could have been diagnosed and treated early, with a 90 percent chance of long term survival.
‘Even though he was in palliative care, I never thought he was going to die,’ Susie says now.
‘I kept telling myself he was a miracle baby. He was such a soldier, he tried so hard – and to know that one simple test could potentially have saved his life was heartbreaking.’
Now though, other parents like Susie and Justin may be spared their grief: last month it emerged that hundreds of newborn babies are being tested for up to 200 rare genetic conditions, among them SCID.
The world-first Generation Study, run in a partnership between Genomics England and NHS England, will see up to 100,000 babies given whole genomic sequencing.
This will provide a readout of a person’s entire genetic code and check for changes linked to specific health conditions — using blood samples typically taken from the umbilical cord shortly after birth.
It’s a huge advance on the current, routine ‘heel prick’ test undertaken at five days old, when a newborn’s heel is pricked to collect a few drops of blood, which is then sent away to be tested for nine rare but serious conditions, among them cystic fibrosis and hypothyroidism (but not SCID).

James was a much wanted half-brother for Ethan and Ollie (pictured), Susie’s then four-year-old twins from a previous relationship

Susie and Justin today have a healthy six-year-old daughter called Katie (pictured) who, as a girl, can only be a carrier of SCID
If deemed a success, the scheme could be rolled out nationwide.
It follows years of campaigning by paediatricians and researchers who were convinced of the vast difference testing could make to the outcomes of children who, while apparently symptomless at birth, went on to develop life-changing — and life-ending — problems.
Among them was Bobby Gaspar, a world-renowned professor in paediatrics and immunology whose years at Great Ormond Street Hospital saw him working on the front line of inherited genetic disease, particularly those that affected the immune system.
‘These babies were born “normally” but after a few months, when their mother’s immunity went away, they couldn’t fight a cough or cold and often the first time we would see them was on a ventilator,’ he recalls.
‘Ultimately, the only way you could treat them was a bone marrow transplant. But regardless of the type of treatment, what we found through all kinds of studies was, because of previous infections, the outcomes were terrible.’
Over the years, he also realised the vast difference in outcomes from first-borns and siblings who had been tested at birth because their genetic risk had been highlighted by their older brothers and sisters.
‘So the first child would present with these problems and outcomes were poor, but if you looked at siblings you could test those babies at birth and find out if they had the same defective genes.
‘And if they did, you could start to protect them from birth with bone marrow transplants and gene therapy — and virtually all of them survived,’ he says.

The first flag that something was wrong came after James’ routine eight week vaccinations
Armed with this knowledge — and the astonishing advances in screening technologies — Bobby and his colleagues spent years campaigning for further compulsory screening for SCID.
It took ten years, but in 2021 their patience and determination was rewarded with a two-year pilot study of blood test screening for SCID at birth.
Simultaneously he was approached to consult on the Generation Study.
‘It’s been fantastic, and long overdue, to see things starting to happen in this area,’ he says.
No-one knows this more than Susie and Justin, who today have a healthy six-year-old daughter called Katie who, as a girl, can only be a carrier of SCID.
Neither could ever have predicted what lay ahead when, in February 2016, a delighted Susie gave birth to their first child together, an apparently healthy 8lbs 8oz baby boy who was a much wanted half-brother for Ethan and Ollie, Susie’s then four-year-old twins from a previous relationship, and Justin’s two older sons Henry, 14, and 12-year-old Joe.
The first flag that something was wrong came after James’ routine eight week vaccinations.
‘In the five minutes it took me to drive home he developed a really horrible cough,’ Susie recalls.

Pictured, James with twins Ethan and Ollie and Justin’s two older sons Henry, 14, and 12-year-old Joe
‘It was really strange and when he still wasn’t quite himself the next day I took him back to the doctors who told me it was just a cold and to keep an eye on him.’
It was a refrain Susie and Justin would hear frequently over the next few weeks, as James seemed to develop one infection after another.
At one point, he was admitted to Norwich and Norfolk hospital for six days and administered intravenous antibiotics for what was deemed to be a chest infection, only to be sent home again.
‘I’d had twins, I knew something still wasn’t right,’ Susie recalls. ‘I went back to the doctors who said chest infections could take a while and to carry on with the oral antibiotics.’
When James stopped eating or drinking Susie once more went to the doctors, who told her his lack of appetite might be down to a case of oral thrush.
‘The doctor said “he looks so well”, but in the end agreed to send him to hospital as his heart rate was so fast.
‘Once we were there, they found his levels were at 47 when they should be 100. At this point all hell broke loose,’ she recalls.
Nonetheless, assorted doctors still continued to maintain it was a chest infection, until, two weeks later, another doctor told a desperate Susie and Justin that James needed to be transferred to a specialist hospital as a matter of urgency: his life now hung in the balance.

Only after a blue light dash to Addenbrookes Hospital in Cambridge were the couple told by intensive care doctors that James had SCID and needed to be transferred to Great Ormond Street
‘This is after two weeks of being told his symptoms were “normal for a chest infection”, says Susie. ‘It was completely surreal.’
Only after a blue light dash to Addenbrookes Hospital in Cambridge were the couple told by intensive care doctors that their son had SCID and needed to be transferred to Great Ormond Street, where in turn they learned that James urgently needed a bone marrow transplant.
With no time to wait to find a full match, Justin donated his T-cells — white blood cells which form part of the immune system.
‘It was then a matter of waiting to see if they would help James start to build an immune system of his own,’ says Susie, who had to juggle the worry of her seriously ill baby son with caring for her young twins.
Tragically, the bad news kept coming.
‘Just as he was improving a little, he started having seizures and was diagnosed with infantile spasms. It felt like one terrible diagnosis after another,’ she says.
Then, just before the end of the year came the worst diagnosis at all, when doctors confirmed that there was no T-cell growth.
James’ body was also riddled with CMV, a common virus which, while not usually harmful, can be devastating to those with a weakened immune system and had left him blind and severely brain damaged.

Ally Shaw and husband Jake are also facing their own bitter sting in the tail too. Both their daughters Nala (left), now five, and three-year-old Teddi (right) have metachromatic leukodystrophy (MLD)

Today Nala is completely wheelchair bound, cannot move her own body or speak, is blind and is fed through a peg in her stomach
James was sent home for palliative care, and died five days before his first birthday in his mother’s arms.
‘Even right at the end it was hard for me to believe it because he was such a fighter,’ she says.
It is a heartbreak that, many miles away from the Thorndyke home, in Northumberland, Ally Shaw and husband Jake are also facing — with their own bitter sting in the tail too.
Both their daughters Nala, now five, and three-year-old Teddi have metachromatic leukodystrophy (MLD), a harrowing genetic disease that causes progressive damage to the affected child’s nervous system.
Few sufferers live beyond their tenth birthday.
Yet because her elder sister had been diagnosed with the condition, Teddi was screened earlier enough to be treated with a new life-saving therapy.
‘The thought that, as a parent, one of your children is going to be saved and one is not, is the worst form of sadness you can have,’ Ally, a 31-year old former care officer says now.
‘The only comfort is telling myself that Nala saved Teddi’s life.’

Still just a baby, Teddi was eligible for a revolutionary new therapy called Libmeldy which works by removing the child’s stem cells and replacing the faulty gene that causes MLD before re-injecting the treated cells into the patient
Today Nala is completely wheelchair bound, cannot move her own body or speak, is blind and is fed through a peg in her stomach, a deterioration that has taken place over the two and a half years since she was diagnosed, and has clearly been incredibly distressing for her parents — who separated earlier this year, in part due to the stress of Nala’s condition — to watch.
Alongside the sorrow is anger too, at the many missed opportunities for Nala to be given a diagnosis.
When as a young toddler, she started to fall over regularly, Ally, 34, and Jake were told by doctors and physios she would ‘grow out of it’.
Even after she developed a tremor in her body, it would take weeks for her anxious parents to secure an appointment with a paediatrician.
At this point they were told she might have cerebral palsy.
‘I struggled to believe that as she was nearly two and a half by then, and I thought it would have been picked up by now,’ Ally says. ‘I was convinced she had a brain tumour.’
Nala was put on a waiting list for an MRI, during which time her condition dramatically worsened.
‘She couldn’t pull herself up on the sofa, she couldn’t get herself off, and at night time, she was screaming in pain, and her whole body was trembling,’ Ally recalls.

Happily Teddi, who began treatment — which is similar to dialysis — in June 2022, aged one, and was subsequently discharged home four months later. Today, more than two years on, she hasn’t displayed symptoms
‘It was horrible to see, so one night in desperation I called an ambulance.’
On arrival at Newcastle, Nala was rushed for an emergency MRI as doctors suspected a brain tumour.
‘For us this was the worst case scenario and when doctors said it wasn’t a tumour, I was almost doing somersaults round the room,’ says Ally.
‘But then they told us they had found evidence of MLD.’
Neither of them had heard of the disease before, but it did not take long to realise what it meant for their much-loved daughter.
‘At first there was a lot of denial,’ Ally says quietly. ‘You tell yourself your child is different, that she will defy the odds.’
At the same time, the couple had to face up to another issue: as MLD is genetic, their younger daughter Teddi, who had been born in June 2021, may have it too.
A week later, after testing, their worst fears were realised when they were told she also carried the faulty gene.
This time though, the prognosis was different.
Still just a baby, Teddi was eligible for a revolutionary new therapy called Libmeldy which works by removing the child’s stem cells and replacing the faulty gene that causes MLD before re-injecting the treated cells into the patient.
‘You have to give it before these terrible problems start,’ says Bobby Gaspar, whose research company Orchard Therapeutics developed the drug.
‘That means the most effective thing you can do is screen at birth.’
For the Shaws meanwhile, the news was a difficult pill to swallow.
‘While we were happy that one of our children would have a chance, at the same time there was lots of anger, because if we had been listened to just a few months earlier Nala would probably would have been eligible for treatment.
‘There were so many missed opportunities,’ Ally says.
Happily Teddi, who began treatment — which is similar to dialysis — in June 2022, aged one, and was subsequently discharged home four months later.
Today, more than two years on, she hasn’t displayed symptoms.
‘She’s bright as a button, walking, talk and running, jumping, all things Nala could never do and will never do,’ says Ally.
Its why, like Susie, she is also a passionate advocate for genomic newborn screening to be offered to all children.
‘It makes no sense not to do it,’ she says. ‘Not doing it means that children who could live a long and healthy life are going to die.’
‘There is no downside,’ adds Bobby.
‘These severe genetic diseases are rare, but cumulatively the burden they impose on the health service is immense.
‘If we can roll this out, it changes the landscape of medicine, because we are now in a position to identify a condition or disease at birth and then do something that means the child never sees the disease, the family doesn’t have to live with the terrible consequences and the NHS doesn’t have to bear the burden of looking after a chronically ill child that is in and out of hospital the whole time.
‘It’s preventative medicine at its best.’



